Actualités
Cette étude observationnelle, présentée fin mars, met en évidence le lien entre l'infection par la...
- Actualités
- Science

Widex a été distingué pour son SmartRic et son accessoire multifonction Sound Assist....
- Actualités
- Évènements
- Fabricants
- Marché
- Produits

Le Conseil de l'Âge du HCFEA se réfère aux conclusions de son rapport « Bien...
- Actualités
- Politique de santé




Si l'immense majorité des audioprothésistes fait un travail sérieux, le dynamisme du secteur a récemment attiré des personnes moins scrupuleuses. Les créations...
- Législation
- Politique de santé

Le 44e Congrès des audioprothésistes a été inauguré par le Pr Lionel Collet, président de la Haute Autorité de Santé (HAS) et François...
- Actualités
- Évènements

Dans cet épisode, Madame Oreille dépanne un patient au fin fond de la montagne et par temps de neige... à ses risques...
- Actualités
- Podcasts
- Vie pro
Magazines

Innovations et produits

Unitron a présenté, mardi 2 avril, Stride V-UP, un contour d’oreille « ultra-puissant » conçu...
- Actualités
- Aides auditives
- Fabricants
- Innovations et produits
- Produits

Hansaton a présenté, mardi 2 avril, les nouvelles aides auditives Hansaton Fokus Ultra Power, ses...
- Actualités
- Aides auditives
- Fabricants
- Innovations et produits
- Produits
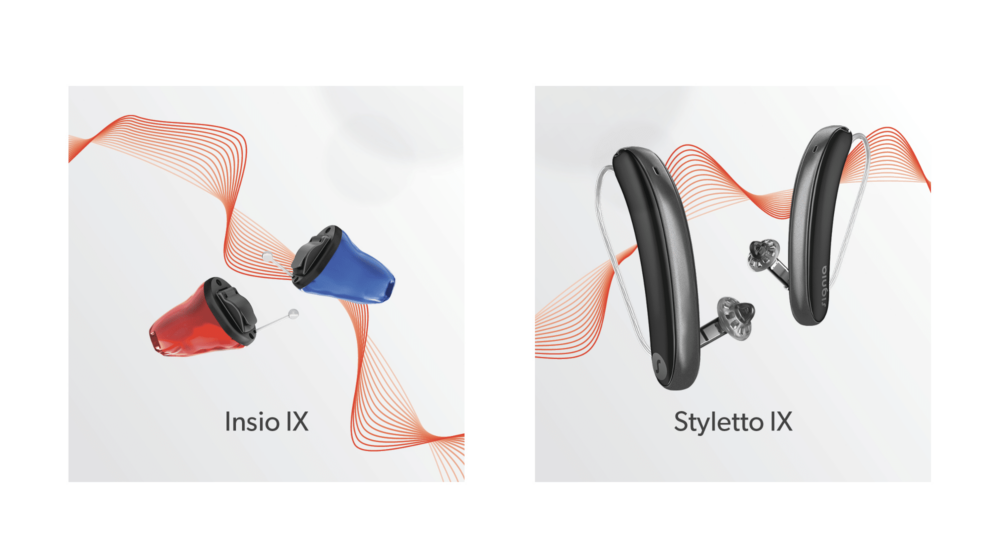
Deux modèles phares de Signia, le Styletto IX et l'Insio IX, sont désormais dotés de...
- Actualités
- Aides auditives
- Fabricants
- Innovations et produits
Vie pro

Le réseau Audilab a lancé la nouvelle saison de son podcast « Appareillons ». Le...
- Actualités
- Enseignes
- Évènements
- Fabricants
- Podcasts
- Vie pro

Jan Makela succède au français Eric Bernard au poste de président directeur général de WS...
- Actualités
- Évènements
- Fabricants
- Marché
- Vie pro

Du 20 au 22 septembre prochain se tiendra le 130e Congrès de la SFORL au...
- Actualités
- Évènements
- Organisations professionnelles
- Politique de santé
- Vie pro
Podcasts
Le réseau Audilab a lancé la nouvelle saison de son podcast « Appareillons ». Le...
- Actualités
- Enseignes
- Évènements
- Fabricants
- Podcasts
- Vie pro
Dans cet épisode, Madame Oreille dépanne un patient au fin fond de la montagne et...
- Actualités
- Podcasts
- Vie pro
Dans cet épisode, Madame Oreille rencontre une série de patients avec des bouchons de cérumen....
- Actualités
- Podcasts
- Vie pro
Dans cet épisode, Madame Oreille s'interroge sur le questionnaire de satisfaction que sont censés remplir...
- Actualités
- Podcasts
- Vie pro
Dans cet épisode, Madame Oreille revient sur sa lecture du rapport de l'IGAS de février...
- Actualités
- Podcasts
- Vie pro
Madame Oreille est audioprothésiste indépendante, elle nous livre avec humour son quotidien : dans cet...
- Actualités
- Podcasts
- Vie pro
Madame Oreille est audioprothésiste indépendante, elle nous livre avec humour son quotidien : dans cet...
- Actualités
- Podcasts
- Vie pro